
起こりうる副作用とその対策
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行ってください。
(1)重大な副作用
1)低カルシウム血症(5.6%※)
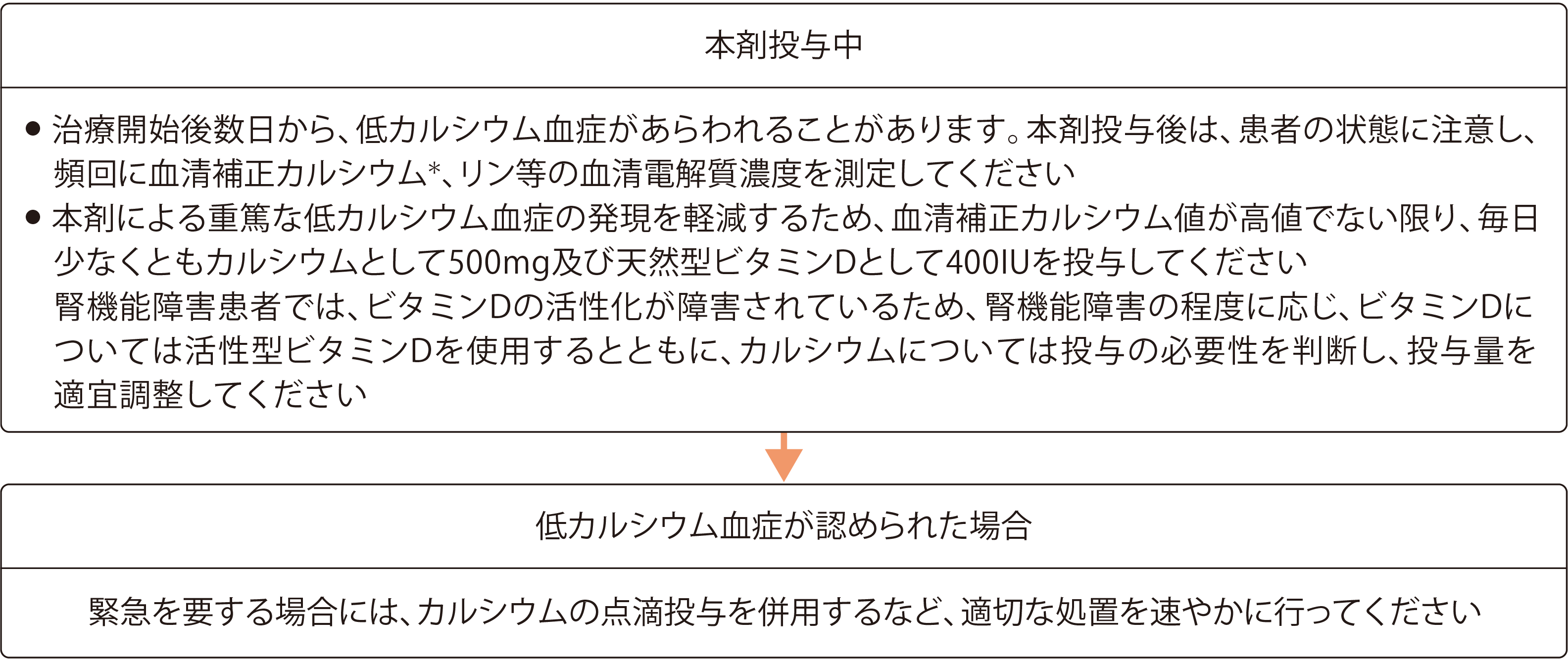
QT延長、痙攣、テタニー、しびれ、失見当識等の症状を伴う低カルシウム血症があらわれることがあり、死亡に至った例が報告されています。低カルシウム血症が認められた場合には、カルシウム及びビタミンDの経口投与に加えて、緊急を要する場合には、カルシウムの点滴投与を併用するなど、適切な処置を速やかに行ってください。
患者様に対し、本剤の投与中に低カルシウム血症が疑われる症状があらわれた場合には、速やかに主治医に連絡するよう指導してください。
※先行バイオ医薬品(ランマーク®)の臨床試験結果に基づく副作用発現頻度
予防
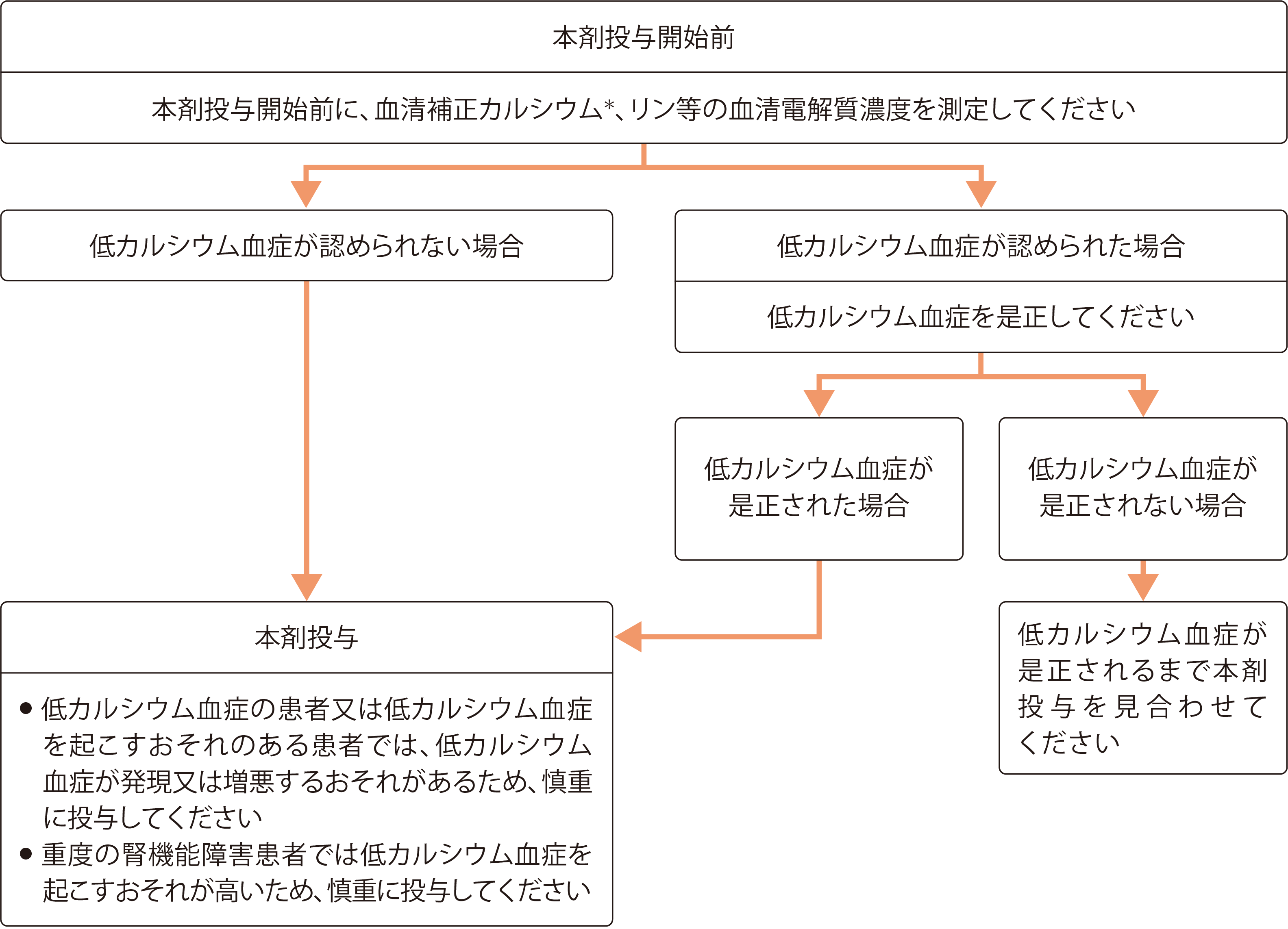
- 本剤の投与開始後数日から、低カルシウム血症が認められることがあります。そのため、本剤の投与開始後は、患者様の状態に注意し、頻回に血清カルシウム、リン等の血清電解質濃度を測定してください。
- 本剤の投与開始後は、血清補正カルシウム値が高値でない限り、毎日少なくともカルシウムとして500mg及び天然型ビタミンDとして400IUを投与してください。
本剤の海外第Ⅲ相臨床試験*1において、本剤60mgが投与された56%の患者様にカルシウムとビタミンD及び/又はその他薬剤の配合錠*2が使用されました。
- 腎機能障害患者様では、ビタミンDの活性化が障害されていますので、腎機能障害の程度に応じ、ビタミンDについては活性型ビタミンDを使用するとともに、カルシウムについては投与の必要性を判断し、投与量を適宜調整してください。
- 「本剤投与開始前及び投与中の低カルシウム血症への対策」及び「本剤投与開始前及び投与中の低カルシウム血症に対するチェック項目」も参考に適切な対応・処置を行ってください。
対処法
- 本剤によるグレード3又は4の低カルシウム血症が発現した場合、グレード1以下に回復するまで休薬を考慮してください(グレードはCTCAEに準じる、低カルシウム血症のCTCAEグレードは「(参考)低カルシウム血症」をご参照ください)。
- 緊急を要する低カルシウム血症の場合には、カルシウムの点滴投与を併用するなど、適切な処置を速やかに行ってください。
*1 試験概要についてはこちらをご参照ください。
*2 本剤と有効成分が同等の医療用医薬品:一般名 沈降炭酸カルシウム/コレカルシフェロール(天然型ビタミンD)/炭酸マグネシウム配合錠についての電子添文の抜粋は以下のとおりである。
4. 効能又は効果 RANKL阻害剤(デノスマブ(遺伝子組換え)等)投与に伴う低カルシウム血症の治療及び予防
6. 用法及び用量 通常、1日1回2錠を経口投与する。なお、患者の状態又は臨床検査値に応じて適宜増減する。
1. 警告(抜粋)
1.3
本剤投与後に低カルシウム血症が認められた場合には、カルシウム及びビタミンDの経口投与に加えて、緊急を要する場合には、カルシウムの点滴投与を併用するなど、適切な処置を速やかに行うこと。[11.1.1参照]
7. 用法及び用量に関連する注意
7.1
本剤によるグレード3又は4の副作用が発現した場合、グレード1以下に回復するまで休薬を考慮すること(グレードはCTCAEに準じる)。
7.2
本剤による重篤な低カルシウム血症の発現を軽減するため、血清補正カルシウム値が高値でない限り、毎日少なくともカルシウムとして500mg及び天然型ビタミンDとして400IUの投与を行うこと。ただし、腎機能障害患者では、ビタミンDの活性化が障害されているため、腎機能障害の程度に応じ、ビタミンDについては活性型ビタミンDを使用するとともに、カルシウムについては投与の必要性を判断し、投与量を適宜調整すること。[1.1、17.1.1-17.1.3参照]
8. 重要な基本的注意(抜粋)
8.3
治療開始後数日から、低カルシウム血症があらわれることがある。本剤投与後は、患者の状態に注意し、頻回に血清カルシウム、リン等の血清電解質濃度を測定すること。[11.1.1参照]
本剤投与開始前及び投与中の低カルシウム血症への対策
低カルシウム血症への対策(本剤投与開始前)
低カルシウム血症への対策(本剤投与中)
*血清アルブミンが4.0g/dL未満の場合には、以下の式による補正カルシウム値を用いる。
補正カルシウム値(mg/dL)=血清カルシウム値(mg/dL)+4-血清アルブミン値(g/dL)
本剤投与開始前及び投与中の低カルシウム血症に対するチェック項目
本剤投与開始前
| チェック項目 | チェック |
|---|---|
投与開始前の血清補正カルシウム、リン等の血清電解質濃度の測定
| □ |
低カルシウム血症を起こすおそれのある患者
| □ |
本剤投与中
| チェック項目 | チェック |
|---|---|
カルシウム及びビタミンDの連日投与
| □ |
投与開始後の血清補正カルシウム、リン等の血清電解質濃度の測定
| □ |
低カルシウム血症が認められた場合の対応
| □ |
患者への指導(低カルシウム血症が疑われる症状があらわれた場合の対応)
| □ |
*血清アルブミンが4.0g/dL未満の場合には、以下の式による補正カルシウム値を用いる。
補正カルシウム値(mg/dL)=血清カルシウム値(mg/dL)+4-血清アルブミン値(g/dL)
(参考)
低カルシウム血症
概要1)
低カルシウム血症は、特異的な自覚症状に乏しく、無症候性であることが多いため発見・診断が遅れることが多い電解質異常であることが知られています。
低カルシウム血症の症状は、軽症では無症状か、筋肉のこわばり程度にとどまります。重症では、全身の筋痙攣(テタニー)、強直性のけいれん、意識消失を起こすことがあります。また、抑うつや不安などの精神症状を伴う場合もあり、精神疾患やてんかんと誤診されることもあります。
低カルシウム血症の主な原因には、副甲状腺機能低下症、ビタミンDの欠乏や代謝異常、腎臓病、薬剤の影響などがあります。これらにより血中カルシウムの調節が障害され、低カルシウム血症が起こります。
診断1)
血清カルシウム値が8.5mg/dL未満の場合を低カルシウム血症と診断します。
血清カルシウム値は、低アルブミン血症などがあると、カルシウム代謝に異常がなくても低値となるため、見かけ上、低カルシウム血症を示すことになります。そのため、血清アルブミンが4.0g/dL 未満の場合には、以下の式による補正カルシウム値を用います。
補正カルシウム値(mg/dL)=血清カルシウム値(mg/dL)+4-血清アルブミン値(g/dL)
また、低カルシウム血症のCTCAEグレード分類2)は以下のとおりです。
| グレード1 | グレード2 | グレード3 | グレード4 | グレード5 | |
| 低カルシウム 血症 | 補正血清カルシウム <LLN-8.0mg/dL; <LLN-2.0mmol/L; イオン化カルシウム <LLN-1.0mmol/L | 補正血清カルシウム <8.0-7.0mg/dL; <2.0-1.75mmol/L; イオン化カルシウム <1.0-0.9mmol/L; 症状がある | 補正血清カルシウム <7.0-6.0mg/dL; <1.75-1.5mmol/L; イオン化カルシウム <0.9-0.8mmol/L; 入院を要する | 補正血清カルシウム <6.0mg/dL; <1.5mmol/L; イオン化カルシウム <0.8mmol/L; 生命を脅かす | 死亡 |
グレードはCTCAE v5.0に基づく。
1)山内美香:日本内科学会雑誌. 2020; 109(4): 733-739より改変
2)有害事象共通用語規準 v5.0 日本語訳JCOG版より引用
2)顎骨壊死・顎骨骨髄炎(1.8%※)
顎骨壊死・顎骨骨髄炎があらわれることがあり、本剤の長期投与により顎骨壊死の発現率の増加が認められています。
本剤の投与中に歯科処置が必要になった場合には、できる限り非侵襲的な歯科処置を受けるよう指導してください。
異常が認められた場合には、直ちに歯科・口腔外科を受診するよう指導してください。
※先行バイオ医薬品(ランマーク®)の臨床試験結果に基づく副作用発現頻度
予防
- 報告された症例の多くが抜歯等の顎骨に対する侵襲的な歯科処置や局所感染に関連して発現しています。
リスク因子としては、悪性腫瘍、化学療法、血管新生阻害薬、コルチコステロイド治療、放射線療法、口腔の不衛生、歯科処置の既往等が知られています。 - 本剤の投与開始前は口腔内の管理状態を確認し、必要に応じて、患者様に対し適切な歯科検査を受け、侵襲的な歯科処置をできる限り済ませておくよう指導してください。
- 本剤の投与中に歯科処置が必要になった場合には、できる限り非侵襲的な歯科処置を受けるよう指導してください。
- また、口腔内を清潔に保つこと、定期的な歯科検査を受けること、歯科受診時に本剤の使用を歯科医師に告知して侵襲的な歯科処置はできる限り避けることなどを患者様に十分説明してください。
対処法
- 本剤によるグレード3又は4の顎骨壊死・顎骨骨髄炎が発現した場合、グレード1以下に回復するまで休薬を考慮してください(グレードはCTCAEに準じる)。
- 本剤の投与中に異常が認められた場合には、直ちに歯科・口腔外科を受診するように指導してください。
(参考)
薬剤関連顎骨壊死(medication-related osteonecrosis of the jaw;MRONJ)の診断と発症メカニズム
MRONJの診断1)
以下の3項目を満たした場合にMRONJと診断する。
- ビスホスホネート(BP)やデノスマブ製剤による治療歴がある。
- 8週間以上持続して、口腔・顎・顔面領域に骨露出を認める。または口腔内、あるいは口腔外から骨を触知できる瘻孔を8週間以上認める。
- 原則として、顎骨への放射線照射歴がない。また顎骨病変が原発性がんや顎骨へのがん転移でない。
MRONJの発症メカニズム1)
これまでの研究成果の蓄積から、MRONJの発症や進展に関わる仮説として以下のメカニズムが考えられている。
- 骨のリモデリング阻害
- 細菌感染
- 血管新生阻害
1)顎骨壊死検討委員会. 薬剤関連顎骨壊死の病態と管理:顎骨壊死検討委員会ポジションペーパー2023, 2023
8. 重要な基本的注意(抜粋)
8.4
顎骨壊死・顎骨骨髄炎があらわれることがあり、本剤の長期投与により顎骨壊死の発現率の増加が認められている。報告された症例の多くが抜歯等の顎骨に対する侵襲的な歯科処置や局所感染に関連して発現している。リスク因子としては、悪性腫瘍、化学療法、血管新生阻害薬、コルチコステロイド治療、放射線療法、口腔の不衛生、歯科処置の既往等が知られている。本剤の投与開始前は口腔内の管理状態を確認し、必要に応じて、患者に対し適切な歯科検査を受け、侵襲的な歯科処置をできる限り済ませておくよう指導すること。
本剤投与中に歯科処置が必要になった場合には、できる限り非侵襲的な歯科処置を受けるよう指導すること。また、口腔内を清潔に保つこと、定期的な歯科検査を受けること、歯科受診時に本剤の使用を歯科医師に告知して侵襲的な歯科処置はできる限り避けることなどを患者に十分説明し、異常が認められた場合には、直ちに歯科・口腔外科を受診するように指導すること。[11.1.2参照]
3)大腿骨、尺骨等の非定型骨折(頻度不明※)
本剤又はビスホスホネート系薬剤を長期使用している患者様において、非外傷性又は軽微な外力による大腿骨転子下、近位大腿骨骨幹部、近位尺骨骨幹部等の非定型骨折が発現したとの報告があります。
本剤の投与開始後にこのような症状が認められた場合には、X線検査等を行い、適切な処置を行ってください。
また、両側性の骨折が生じる可能性があることから、片側で非定型骨折が起きた場合には、反対側の部位の症状等を確認し、X線検査を行うなど、慎重に観察してください。
※先行バイオ医薬品(ランマーク®)の臨床試験結果に基づく副作用発現頻度
予防
- 本剤又はビスホスホネート系薬剤を長期使用している患者様において、非外傷性又は軽微な外力による大腿骨転子下、近位大腿骨骨幹部、近位尺骨骨幹部等の非定型骨折が発現したとの報告があります。
これらの報告では、完全骨折が起こる数週間から数ヵ月前に大腿部、鼠径部、前腕部等において前駆痛が認められている報告もあることから、本剤の投与開始後にこのような症状が認められた場合には、X線検査等を行い、適切な処置を行ってください。 - 両側性の骨折が生じる可能性があることから、片側で非定型骨折が起きた場合には、反対側の部位の症状等を確認し、X線検査を行うなど、慎重に観察してください。
対処法
- 本剤によるグレード3又は4の大腿骨、尺骨等の非定型骨折が発現した場合、グレード1以下に回復するまで休薬を考慮してください(グレードはCTCAEに準じる)。
- X線検査時に骨皮質の肥厚等、特徴的な画像所見が認められた場合には適切な処置を行ってください。
4)アナフィラキシーを含む過敏症(頻度不明※)
本剤の投与により過敏症があらわれることがあります。過敏症による症状が認められた場合には適切な処置を行ってください。
※先行バイオ医薬品(ランマーク®)の臨床試験結果に基づく副作用発現頻度
対処法
- 本剤の投与により過敏症に関連する症状があらわれることがあります(「本剤投与時に観察すること」参照)。そのため、十分な観察を行ってください。過敏症に関連する症状が認められた場合には適切な処置を行ってください。
- 本剤によるグレード3又は4のアナフィラキシーを含む過敏症が発現した場合、グレード1以下に回復するまで休薬を考慮してください(グレードはCTCAEに準じる)。
8. 重要な基本的注意(抜粋)
8.5
本剤又はビスホスホネート系薬剤を長期使用している患者において、非外傷性又は軽微な外力による大腿骨転子下、近位大腿骨骨幹部、近位尺骨骨幹部等の非定型骨折が発現したとの報告がある。これらの報告では、完全骨折が起こる数週間から数ヵ月前に大腿部、鼠径部、前腕部等において前駆痛が認められている報告もあることから、本剤の投与開始後にこのような症状が認められた場合には、X線検査等を行い、適切な処置を行うこと。また、両側性の骨折が生じる可能性があることから、片側で非定型骨折が起きた場合には、反対側の部位の症状等を確認し、X線検査を行うなど、慎重に観察すること。X線検査時には骨皮質の肥厚等、特徴的な画像所見がみられており、そのような場合には適切な処置を行うこと。[11.1.4参照]
5)治療中止後の多発性椎体骨折(頻度不明※)
本剤の投与中止後に、多発性椎体骨折があらわれることがあります。本剤による治療を中止する場合には、骨吸収抑制薬の使用を考慮してください。
※先行バイオ医薬品(ランマーク®)の臨床試験結果に基づく副作用発現頻度
対処法
- 本剤の投与中止後には、骨吸収が一過性に亢進し、多発性椎体骨折があらわれることがあります。そのため、投与を中止する場合には、本剤の投与中止後に骨吸収抑制薬(ビスホスホネート製剤等)の使用を考慮してください。
3. 組成・性状(抜粋)3.1 組成 販売名:デノスマブBS皮下注120mgRM「F」、有効成分:1バイアル(1.7mL)中デノスマブ(遺伝子組換え)注)[デノスマブ後続1]120mg、添加剤:1バイアル(1.7mL)中L-ヒスチジン 0.524mg、L-ヒスチジン塩酸塩水和物 1.07mg、精製白糖 140mg、ポリオキシエチレン(160)ポリオキシプロピレン(30)グリコール 0.51mg
注)本剤は遺伝子組換え技術によりチャイニーズハムスター卵巣(CHO)細胞を用いて製造される。
4. 効能又は効果
多発性骨髄腫による骨病変及び固形癌骨転移による骨病変
6. 用法及び用量
通常、成人にはデノスマブ(遺伝子組換え)[デノスマブ後続1]として120mgを4週間に1回、皮下投与する。
(2)副作用一覧
本試験には承認外の効能又は効果、用法及び用量の情報が含まれているが、本剤と先行バイオ医薬品 (US)の60mg製剤*との有効性の同等性を検討した承認時評価資料のため紹介する。
*先行バイオ医薬品(US)の60mg製剤は米国で承認されているプラリア®〔デノスマブ(遺伝子組換え)製剤〕を指す。
〈海外第Ⅲ相臨床試験(AVT03-GL-C01試験、海外データ)〉
1)社内資料:海外第Ⅲ相臨床試験成績(AVT03-GL-C01)[承認時評価資料]
2)Lortkipanidze M, et al.: Expert Opin Biol Ther. 2025: 25(8): 899-912
(COI:本試験はAlvotech社の資金提供により実施された。著者の中にAlvotech社の社員が含まれる)
1)12ヵ月時点までの副作用等
副作用は本剤60mg群では266例中59例(22.2%)、先行バイオ医薬品群では266例中50例(18.8%)に認められた。主な副作用(3%以上に発現)は、本剤60mg群では低カルシウム血症24例(9.0%)、補正カルシウム減少11例(4. 1%)、注射部位反応9例(3. 4%)、先行バイオ医薬品群では低カルシウム血症18例(6. 8%)、注射部位反応8例(3. 0%)であった。
副作用(安全性解析対象集団)
| 本剤60mg群 (n=266) | 先行バイオ医薬品群 (n=266) | |
| n(%) | n(%) | |
| 副作用発現例 | 59(22.2) | 50(18.8) |
| 代謝および栄養障害 | 24(9.0) | 19(7.1) |
| 低カルシウム血症 | 24(9.0) | 18(6.8) |
| 高カルシウム血症 | 0 | 1(0.4) |
| 筋骨格系および結合組織障害 | 13(4.9) | 17(6.4) |
| 筋骨格痛 | 6(2.3) | 7(2.6) |
| 関節痛 | 3(1.1) | 4(1.5) |
| 四肢痛 | 3(1.1) | 2(0.8) |
| 筋肉痛 | 1(0.4) | 2(0.8) |
| 脊椎痛 | 1(0.4) | 1(0.4) |
| 背部痛 | 0 | 1(0.4) |
| 関節硬直 | 1(0.4) | 0 |
| 関節腫脹 | 1(0.4) | 0 |
| 筋痙縮 | 0 | 1(0.4) |
| 頚部痛 | 1(0.4) | 0 |
| 滑液嚢腫 | 0 | 1(0.4) |
| 臨床検査 | 18(6.8) | 9(3.4) |
| 補正カルシウム減少 | 11(4.1) | 3(1.1) |
| 活性化部分トロンボプラスチン時間延長 | 6(2.3) | 2(0.8) |
| カルシウムイオン減少 | 3(1.1) | 3(1.1) |
| 補正カルシウム増加 | 0 | 1(0.4) |
| 国際標準比増加 | 0 | 1(0.4) |
| 一般・全身障害および投与部位の状態 | 12(4.5) | 8(3.0) |
| 注射部位反応 | 9(3.4) | 8(3.0) |
| 疲労 | 2(0.8) | 0 |
| 倦怠感 | 1(0.4) | 0 |
| 末梢性浮腫 | 1(0.4) | 0 |
| 皮膚および皮下組織障害 | 2(0.8) | 1(0.4) |
| 脱毛症 | 1(0.4) | 1(0.4) |
| アレルギー性皮膚炎 | 1(0.4) | 0 |
| 胃腸障害 | 1(0.4) | 1(0.4) |
| アフタ性潰瘍 | 1(0.4) | 0 |
| 便秘 | 0 | 1(0.4) |
| 神経系障害 | 1(0.4) | 1(0.4) |
| 浮動性めまい | 1(0.4) | 0 |
| 頭痛 | 0 | 1(0.4) |
| 血液およびリンパ系障害 | 1(0.4) | 0 |
| 凝固因子異常 | 1(0.4) | 0 |
| 耳および迷路障害 | 0 | 1(0.4) |
| 耳鳴 | 0 | 1(0.4) |
| 肝胆道系障害 | 1(0.4) | 0 |
| 胆石症 | 1(0.4) | 0 |
| 腎および尿路障害 | 1(0.4) | 0 |
| 非感染性膀胱炎 | 1(0.4) | 0 |
| 呼吸器、胸郭および縦隔障害 | 1(0.4) | 0 |
| 呼吸困難 | 1(0.4) | 0 |
| 血管障害 | 1(0.4) | 0 |
| 低血圧 | 1(0.4) | 0 |
MedDRA v27.1
また、重篤な有害事象は本剤60mg群では9例(3.4%)、先行バイオ医薬品群では10例(3.8%)に認められた。内訳は、本剤60mg群では慢性胃炎、びらん性十二指腸炎、膵炎、感染性筋炎、肺炎、軟部組織感染、遠隔転移を伴う肺癌、慢性冠症候群、心室性不整脈、死亡、原発性副甲状腺機能亢進症、脱水、腎不全、性器脱各1例(0.4%)、先行バイオ医薬品群では腹痛、虫垂炎、女性乳癌、骨血管腫、突然死、抑うつ気分、統合失調感情障害、高血圧、腸骨動脈閉塞、頭蓋脳損傷各1例(0.4%)であった。
早期試験中止又は投与中止に至った有害事象は本剤60mg群では8例(3.0%)、先行バイオ医薬品群では3例(1.1%)に認められた。内訳は、本剤60mg群では関節痛、顎痛、脊椎痛、死亡、原発性副甲状腺機能亢進症、糸球体濾過率減少、腎不全、脱毛症各1例(0.4%)、先行バイオ医薬品群では突然死、過敏症、顎膿瘍各1例(0.4%)であった。
死亡に至った有害事象は本剤60mg群では3例(1.1%)、先行バイオ医薬品群では1例(0.4%)に認められた。内訳は、本剤60mg群では肺炎、遠隔転移を伴う肺癌、死亡(原因不明)各1例(0.4%)、先行バイオ医薬品群では突然死1例(0.4%)であった。
2)12ヵ月時点から18ヵ月時点までの副作用等
副作用は本剤60mg群では242例中16例(6.6%)、先行バイオ医薬品-本剤群では122例中4例(3.3%)、先行バイオ医薬品-継続群では122例中5例(4.1%)に認められた。主な副作用(1%以上に発現)は、本剤60mg群では補正カルシウム減少8例(3.3%)、カルシウムイオン減少5例(2.1%)、先行バイオ医薬品-本剤群では補正カルシウム減少2例(1.6%)、先行バイオ医薬品-継続群ではカルシウムイオン減少2例(1.6%)であった。
副作用(安全性解析対象集団)
| 本剤60mg群 (n=242) | 先行バイオ医薬品 -本剤群 (n=122) | 先行バイオ医薬品 -継続群 (n=122) | |
| n(%) | n(%) | n(%) | |
| 副作用発現例 | 16 (6.6) | 4 (3.3) | 5 (4.1) |
| 臨床検査 | 11 (4.5) | 3 (2.5) | 3 (2.5) |
| 補正カルシウム減少 | 8 (3.3) | 2 (1.6) | 0 |
| カルシウムイオン減少 | 5 (2.1) | 1 (0.8) | 2 (1.6) |
| 活性化部分トロンボプラスチン時間延⻑ | 0 | 0 | 1 (0.8) |
| 一般・全身障害および投与部位の状態 | 1 (0.4) | 1 (0.8) | 1 (0.8) |
| 注射部位反応 | 1 (0.4) | 1 (0.8) | 1 (0.8) |
| 代謝および栄養障害 | 2 (0.8) | 0 | 1 (0.8) |
| 低カルシウム血症 | 2 (0.8) | 0 | 1 (0.8) |
| 筋骨格系および結合組織障害 | 1 (0.4) | 0 | 1 (0.8) |
| 骨痛 | 0 | 0 | 1 (0.8) |
| 顎痛 | 1 (0.4) | 0 | 0 |
| 神経系障害 | 1 (0.4) | 0 | 0 |
| 感覚鈍麻 | 1 (0.4) | 0 | 0 |
| 皮膚および皮下組織障害 | 1 (0.4) | 0 | 0 |
| 脱毛症 | 1 (0.4) | 0 | 0 |
MedDRA v27.1
また、重篤な有害事象は本剤60mg群で6例(2.5%)、先行バイオ医薬品-本剤群で3例(2.5%)、先行バイオ医薬品-継続群で2例(1.6%)に認められた。内訳は、本剤60mg群では汎血球減少症、急性腎盂腎炎、前腕骨折、複視、腸閉塞、突然死各1例(0.4%)、先行バイオ医薬品-本剤群では鉄欠乏性貧血、大腿骨骨折、両耳難聴各1例(0.8%)、先行バイオ医薬品-継続群では虫垂炎、うっ血性心不全、胆嚢炎各1例(0.8%)であった。
早期試験中止又は投与中止に至った有害事象はいずれの群においても認められなかった。
死亡に至った有害事象は本剤60mg群では突然死が1例(0.4%)に認められた。先行バイオ医薬品-本剤群、先行バイオ医薬品-継続群では死亡に至った有害事象は認められなかった。
3. 組成・性状(抜粋)3.1 組成 販売名:デノスマブBS皮下注120mgRM「F」、有効成分:1バイアル(1.7mL)中デノスマブ(遺伝子組換え)注)[デノスマブ後続1]120mg、添加剤:1バイアル(1.7mL)中L-ヒスチジン 0.524mg、L-ヒスチジン塩酸塩水和物 1.07mg、精製白糖 140mg、ポリオキシエチレン(160)ポリオキシプロピレン(30)グリコール 0.51mg
注)本剤は遺伝子組換え技術によりチャイニーズハムスター卵巣(CHO)細胞を用いて製造される。
4. 効能又は効果
多発性骨髄腫による骨病変及び固形癌骨転移による骨病変
6. 用法及び用量
通常、成人にはデノスマブ(遺伝子組換え)[デノスマブ後続1]として120mgを4週間に1回、皮下投与する。
3)試験概要
本剤と先行バイオ医薬品の臨床的同等性を比較するための臨床試験に関して、ランマーク®の有する効能又は効果の対象疾患では、併用薬、疾患の状態や病期など の交絡因子により、治療反応の正確な評価が困難であること、また、以下の1)~3)の要因により、対象として、デノスマブを有効成分とするプラリア®の効能又は 効果である骨粗鬆症のうち、閉経後女性患者が感度の高い集団であると考えられた。
- プラリア®の有する効能又は効果である骨粗鬆症では、男性と比較して、骨粗鬆症の閉経後女性でデノスマブの治療効果がより高かった3,4)。ステロイド性骨粗鬆症では、併用薬、疾患の状態や病期などの交絡因子により、治療反応の正確な評価が困難である。
- 骨粗鬆症を有する閉経後女性における骨密度増加に対するデノスマブの有効性は十分に確立されており、椎骨骨折、非椎骨骨折及び大腿骨近位部骨折のリスクを有意に低下させることが示されている5-7)。
- 閉経後女性の骨粗鬆症の有病率は他の効能又は効果の疾患と比較して高いことから、必要な期間内に適切な患者数を確保できる可能性が高いと考えられた8-10)。
以上から、本剤と先行バイオ医薬品の臨床的同等性を比較するための臨床試験では、骨粗鬆症の閉経後女性患者を対象に、本剤の60mg製剤と米国で承認されているプラリア®の臨床的同等性を比較した。
| 目的 | 50歳以上の閉経後骨粗鬆症患者を対象に、本剤の先行バイオ医薬品(US)の60mg製剤*1に対する12ヵ月時点での腰椎骨密度のベースラインからの変化率の同等性について評価する。 | ||||
|---|---|---|---|---|---|
| 試験デザイン | ランダム化、二重盲検、反復投与、2群並行、群間比較、多施設共同試験 | ||||
| 対象 | 50歳以上の閉経後骨粗鬆症患者(女性)532例 | ||||
| 試験方法 | 対象を、閉経からの経過年数及び骨粗鬆症に対する生物学的製剤による治療歴を層別因子として層別し、本剤60mg群(本剤60mgを初回投与し、その後は6及び12ヵ月目に皮下投与)又は先行バイオ医薬品群〔先行バイオ医薬品(US)の60mg製剤を初回投与し、その後は6ヵ月目に皮下投与〕のいずれかに1:1の割合でランダムに割り付けた。先行バイオ医薬品群では同様の層別因子により層別し、先行バイオ医薬品-本剤群(12ヵ月目に本剤60mgを皮下投与)又は先行バイオ医薬品-継続群〔12ヵ月目に先行バイオ医薬品(US)の60mg製剤を皮下投与〕のいずれかに1:1の割合でランダムに割り付けた。 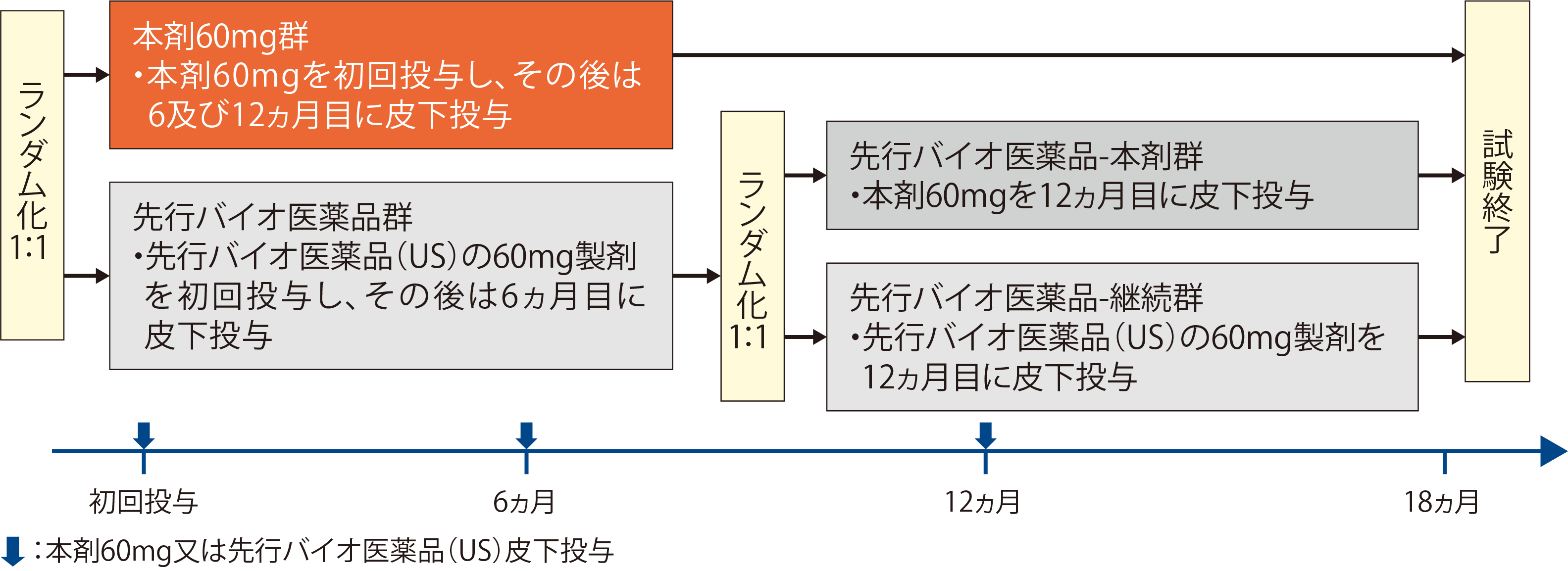 | ||||
| 主要評価項目 | [有効性] 12ヵ月時点での腰椎骨密度のベースラインからの変化率[検証的解析項目] | ||||
| 副次評価項目 | [有効性]
[安全性]
[免疫原性]
[薬物動態(PK)]
[薬力学(PD)]
| ||||
| 解析計画 | [有効性の解析]主要評価項目は、治験薬を1回以上投与したすべての患者を含む最大の解析対象集団(FAS)を対象として解析した。
[安全性の解析] 有害事象*3は、安全性解析対象集団*4を対象として解析し、有害事象の発現患者数及び割合を要約した。 [免疫原性の解析] 安全性解析対象集団で、抗薬物抗体(ADA)及び中和抗体(nAb)の有無を投与群別、治験期間別に集計した。 [PK・PDの解析] PKの解析はPK解析対象集団*5を対象とした。 治療期間別に血液検体を採取し、本剤60mg群及び先行バイオ医薬品群の血清中デノスマブ濃度を評価した。 血清中トラフ濃度は、180日目、12ヵ月目の投与前に採取した検体により解析を実施した。 PDの解析は、PD解析対象集団*6を対象とした。 %Cfb sCTX-1のAUEC0-6monthsの解析は、治療差の過小評価につながる可能性のある中間事象が認められた患者を解析から除外した。 | ||||
| 試験期間 | 2022年8月23日(最初の患者の初回来院)~2024年10月28日(最後の患者の最終来院) |
*1 先行バイオ医薬品(US)の60mg製剤は米国で承認されているプラリア®〔デノスマブ(遺伝子組換え)製剤〕を指す。
*2 ICE:6ヵ月以前に治験薬投与を中止、12ヵ月以前に主要評価項目の評価に影響を及ぼす併用禁止薬を使用、ランダム化された治験薬ではなく、誤った治験薬投与を受けた、主要評価項目の評価に影響を及ぼす治験実施計画書からの逸脱と定義した。
*3 各有害事象はMedDRA v27.1に基づく。
*4 治験薬を1回以上投与したすべての患者
*5 ランダム化されたすべての患者のうち、投与量にかかわらず、1回以上の評価可能なPKパラメータを有する患者
*6 ランダム化されたすべての患者のうち、投与量にかかわらず、1回以上の評価可能なPDパラメータを有する患者
3)Bolognese MA, et al.: J Clin Densitom. 2013; 16(2): 147-153
4)Orwoll E, et al.: J Clin Endocrinol Metab. 2012; 97(9): 3161-3169
5)Bone HG, et al.: J Clin Endocrinol Metab. 2008; 93(6): 2149-2157
6)Cummings SR, et al.: N Engl J Med. 2009; 361(8): 756-765
7)McClung MR, et al.: N Engl J Med. 2006; 354(8): 821-831
8)Meltone 3rd LJ, et al.: J Bone Miner Res. 1992; 7(9): 1005-1010
9)Agrawal AC, et al.: Indian J Orthop. 2023; 57(Suppl1): 45-48
10)Kanis JA, et al.: Osteoporos Int. 2019; 30(1): 3-44
3. 組成・性状(抜粋)3.1 組成 販売名:デノスマブBS皮下注120mgRM「F」、有効成分:1バイアル(1.7mL)中デノスマブ(遺伝子組換え)注)[デノスマブ後続1]120mg、添加剤:1バイアル(1.7mL)中L-ヒスチジン 0.524mg、L-ヒスチジン塩酸塩水和物 1.07mg、精製白糖 140mg、ポリオキシエチレン(160)ポリオキシプロピレン(30)グリコール 0.51mg
注)本剤は遺伝子組換え技術によりチャイニーズハムスター卵巣(CHO)細胞を用いて製造される。
4. 効能又は効果
多発性骨髄腫による骨病変及び固形癌骨転移による骨病変
6. 用法及び用量
通常、成人にはデノスマブ(遺伝子組換え)[デノスマブ後続1]として120mgを4週間に1回、皮下投与する。
